
Akut lymfoblastisk leukæmi - Acute lymphoblastic leukemia
| Akut lymfoblastisk leukæmi | |
|---|---|
| Andre navne | Akut lymfatisk leukæmi, akut lymfoid leukæmi |
 | |
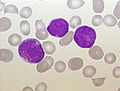
| Udstødning af knoglemarv fra en person med forløber B-celle ALL. De store lilla celler er lymfoblaster. | |
| Specialitet | Hæmatologi , onkologi |
| Symptomer | Trætthed, bleg farve, feber, let blødning eller blå mærker, knoglesmerter, forstørrede lymfeknuder |
| Komplikationer | Infektion , tumorlysesyndrom |
| Almindelig start | 2–5 år gammel |
| Typer | B-celle ALL , T-celle ALL |
| Årsager | Normalt ukendt |
| Risikofaktorer | Enægget tvilling med ALLE, Downs syndrom , Fanconi anæmi , ataksi telangiectasia , Klinefelters syndrom , høj fødselsvægt , betydelig stråling eksponering |
| Diagnostisk metode | Blodprøver og knoglemarvsundersøgelse |
| Differential diagnose | Infektiøs mononukleose , akut myeloid leukæmi , lymfoblastisk lymfom , aplastisk anæmi |
| Behandling | Kemoterapi , stamcelletransplantation , strålebehandling , målrettet terapi |
| Prognose |
Børn : 90% femårig overlevelse Voksne : 35% femårig overlevelse |
| Frekvens | 1 ud af 1.750 børn |
| Dødsfald: Døde | 111.000 (2015) |
Akut lymfoblastisk leukæmi ( ALL ) er en kræft i lymfoidlinjen af blodlegemer karakteriseret ved udvikling af et stort antal umodne lymfocytter . Symptomerne kan være træt, lys hudfarve, feber , let blødning eller blå mærker, forstørrede lymfeknuder eller knoglesmerter. Som en akut leukæmi udvikler ALL sig hurtigt og er typisk dødelig inden for uger eller måneder, hvis den ikke behandles.
I de fleste tilfælde er årsagen ukendt. Genetiske risikofaktorer kan omfatte Downs syndrom , Li-Fraumeni syndrom eller neurofibromatose type 1 . Miljømæssige risikofaktorer kan omfatte betydelig stråleeksponering eller tidligere kemoterapi . Beviser vedrørende elektromagnetiske felter eller pesticider er uklare. Nogle antager, at et unormalt immunrespons på en almindelig infektion kan være en udløser. Den underliggende mekanisme involverer flere genetiske mutationer, der resulterer i hurtig celledeling . De overdrevne umodne lymfocytter i knoglemarven forstyrrer produktionen af nye røde blodlegemer , hvide blodlegemer og blodplader . Diagnosen er typisk baseret på blodprøver og knoglemarvsundersøgelse .
ALL behandles typisk indledningsvis med kemoterapi, der sigter mod at få remission . Dette efterfølges derefter af yderligere kemoterapi typisk over en årrække. Behandlingen omfatter normalt også intratekal kemoterapi, da systemisk kemoterapi kan have begrænset penetration i centralnervesystemet, og centralnervesystemet er et almindeligt sted for tilbagefald af akut lymfoblastisk leukæmi.
Behandlingen kan også omfatte strålebehandling, hvis der er sket spredning til hjernen . Stamcelletransplantation kan anvendes, hvis sygdommen gentager sig efter standardbehandling. Yderligere behandlinger, såsom kimær antigenreceptor T -celle immunterapi , anvendes og undersøges yderligere.
ALLE ramte omkring 876.000 mennesker globalt i 2015 og resulterede i omkring 111.000 dødsfald. Det forekommer hyppigst hos børn, især dem mellem to og fem år. I USA er det den mest almindelige årsag til kræft og død af kræft blandt børn. ALL er kendt for at være den første spredte kræft, der blev helbredt. Overlevelse for børn steg fra under 10% i 1960'erne til 90% i 2015. Overlevelsesraterne er fortsat lavere for babyer (50%) og voksne (35%). Ifølge National Cancer Intelligence Network (NCIN), generelt for mennesker med ALLE: omkring 70 ud af 100 mennesker (70%) vil overleve deres leukæmi i 5 år eller mere, efter at de er blevet diagnosticeret.
tegn og symptomer
Indledende symptomer kan være uspecifikke, især hos børn. Over 50%af børn med leukæmi havde en eller flere af fem funktioner: en lever man kan mærke (64%), en milt man kan mærke (61%), bleg teint (54%), feber (53%) og blå mærker (52%). Derudover kan tilbagevendende infektioner, træthedsfølelse, smerter i arme eller ben og forstørrede lymfeknuder være fremtrædende træk. De B-symptomer , såsom feber, nattesved og vægttab, er ofte til stede som godt.
Symptomer på centralnervesystemet (CNS), såsom kraniale neuropatier på grund af meningeal infiltration, identificeres hos mindre end 10% af voksne og mindre end 5% af børn, især modne B-celle ALL (Burkitt leukæmi) ved præsentation.
Tegn og symptomer på ALL er variable og omfatter:
- Generaliseret svaghed og trætthed
- Anæmi
- Svimmelhed
- Hovedpine, opkastning, sløvhed, nakkestivhed eller kranialnerven parese (CNS involvering)
- Hyppig eller uforklarlig feber og infektion
- Vægttab og/eller tab af appetit
- Overdreven og uforklarlig blå mærker
- Knoglesmerter, ledsmerter (forårsaget af spredning af "blast" -celler til knoglens overflade eller ind i leddet fra marvhulen)
- Åndenød
- Forstørrede lymfeknuder, lever og/eller milt
- Pitting ødem (hævelse) i underekstremiteterne og/eller underlivet
- Petekkier, der er små røde pletter eller linier i huden på grund af lavt blodplade niveauer
- Testikelforstørrelse
- Mediastinal masse
årsag

Kræftcellen i ALL er lymfoblasten. Normale lymfoblaster udvikler sig til modne, infektionsbekæmpende B-celler eller T-celler, også kaldet lymfocytter . Signaler i kroppen styrer antallet af lymfocytter, så hverken der laves for få eller for mange. I ALLE bliver både den normale udvikling af nogle lymfocytter og kontrollen over antallet af lymfoide celler defekte.
ALT dukker op, når en enkelt lymfoblast får mange mutationer til gener, der påvirker blodcelleudvikling og spredning. I barndommen ALL begynder denne proces ved undfangelsen med arv fra nogle af disse gener. Disse gener øger igen risikoen for, at der vil forekomme flere mutationer i udviklingen af lymfoide celler. Visse genetiske syndromer, som Downs syndrom , har samme effekt. Miljørisikofaktorer er også nødvendige for at hjælpe med at skabe nok genetiske mutationer til at forårsage sygdom. Bevis for miljøets rolle ses i barndommen ALL blandt tvillinger, hvor kun 10-15% af begge genetisk identiske tvillinger får ALT. Da de har de samme gener, forklarer forskellige miljøeksponeringer, hvorfor den ene tvilling får ALT og den anden ikke.
Infant ALL er en sjælden variant, der forekommer hos babyer under et år. KMT2A (tidligere MLL ) genarrangementer er mest almindelige og forekommer i embryoet eller fosteret før fødslen. Disse omlejringer resulterer i øget ekspression af blodcelleudviklingsgener ved at fremme gentranskription og gennem epigenetiske ændringer. I modsætning til ALL i barndommen menes miljøfaktorer ikke at spille en væsentlig rolle. Bortset fra KMT2A -omlægningen findes typisk kun en ekstra mutation. Miljøeksponering er ikke nødvendig for at hjælpe med at skabe flere mutationer.
Risikofaktorer
Genetik
Almindelige arvelige risikofaktorer omfatter mutationer i ARID5B , CDKN2A / 2B , CEBPE , IKZF1 , GATA3 , PIP4K2A og mere sjældent TP53 . Disse gener spiller vigtige roller i cellulær udvikling, spredning og differentiering. Individuelt er de fleste af disse mutationer lav risiko for ALLE. Betydelig risiko for sygdom opstår, når en person arver flere af disse mutationer sammen.
Den ujævne fordeling af genetiske risikofaktorer kan hjælpe med at forklare forskelle i sygdomsrater blandt etniske grupper. For eksempel er ARID5B -mutationen mindre almindelig i etniske afrikanske befolkninger.
Flere genetiske syndromer har også øget risiko for ALL. Disse omfatter: Downs syndrom , Fanconi anæmi , Bloom syndrom , X-bundet agammaglobulinæmi , alvorlig kombineret immundefekt , Shwachman-Diamond syndrom , Kostmann syndrom , neurofibromatose type 1 , ataksi-telangiectasia , paroxysmal natlig hæmoglobinuri og Li-Fraumeni syndrom . Færre end 5% af tilfældene er forbundet med et kendt genetisk syndrom.
Sjældne mutationer i ETV6 og PAX5 er forbundet med en familiær form for ALLE med autosomale dominante mønstre af arv .
Miljø
De miljøeksponeringer, der bidrager til fremkomsten af ALL, er omstridte og genstand for løbende debat.
Høje niveauer af strålingseksponering fra nuklear nedfald er en kendt risikofaktor for udvikling af leukæmi. Bevis for, om mindre stråling, som fra røntgenbilleder under graviditet, øger risikoen for sygdom, er fortsat usikker. Undersøgelser, der har identificeret en sammenhæng mellem røntgenbilleder under graviditet og ALL, fandt kun en lidt øget risiko. Eksponering for stærk elektromagnetisk stråling fra kraftledninger har også været forbundet med en lidt øget risiko for ALL. Dette resultat stilles spørgsmålstegn ved, da der ikke kendes nogen årsagsmekanisme, der forbinder elektromagnetisk stråling med kræft.
Høj fødselsvægt (større end 4000 g eller 8,8 lbs) er også forbundet med en lille øget risiko. Mekanismen, der forbinder høj fødselsvægt med ALL, er heller ikke kendt.
Beviser tyder på, at sekundær leukæmi kan udvikle sig hos personer behandlet med visse former for kemoterapi, såsom epipodophyllotoxiner og cyclophosphamid .
Infektioner
Der er nogle tegn på, at en almindelig infektion, såsom influenza , indirekte kan fremme fremkomsten af ALL. Forsinket infektionshypotese siger, at ALLE skyldes et unormalt immunrespons på infektion hos en person med genetiske risikofaktorer. Forsinket udvikling af immunsystemet på grund af begrænset sygdomseksponering kan resultere i overdreven produktion af lymfocytter og øget mutationshastighed under en sygdom. Flere undersøgelser har identificeret lavere satser for ALLE blandt børn med større udsættelse for sygdom tidligt i livet. Meget små børn, der går i daginstitutioner, har lavere satser på ALT. Bevis fra mange andre undersøgelser, der kigger på sygdomseksponering og ALL, er uafklarende. Nogle forskere har knyttet hygiejnehypotesen .
Mekanisme
Flere karakteristiske genetiske ændringer fører til dannelsen af en leukæmisk lymfoblast. Disse ændringer omfatter kromosomale translokationer , intrachromosomale omlejringer , ændringer i antallet af kromosomer i leukæmiske celler og yderligere mutationer i individuelle gener. Kromosomale translokationer involverer at flytte et stort område af DNA fra et kromosom til et andet. Dette skridt kan resultere i placering af et gen fra et kromosom, der fremmer celledeling til et mere aktivt transskriberet område på et andet kromosom. Resultatet er en celle, der deler sig oftere. Et eksempel på dette omfatter translokationen af C-MYC , et gen, der koder for et transkriptionsfaktor , som fører til øget celledeling, ved siden af immunglobulin tung - eller let kæde gen enhancere , hvilket fører til øget C-MYC- ekspression og forøget celledeling . Andre store ændringer i kromosomstruktur kan resultere i placering af to gener direkte ved siden af hinanden. Resultatet er kombinationen af to normalt adskilte proteiner til et nyt fusionsprotein . Dette protein kan have en ny funktion, der fremmer udviklingen af kræft. Eksempler på dette inkluderer ETV6 - RUNX1 -fusionsgenet, der kombinerer to faktorer, der fremmer blodcelleudvikling og BCR - ABL1 -fusionsgenet i Philadelphia -kromosomet . BCR - ABL1 koder for en altid aktiveret tyrosinkinase, der forårsager hyppig celledeling. Disse mutationer producerer en celle, der deler sig oftere, selv i mangel af vækstfaktorer .
Andre genetiske ændringer i B-celle ALL omfatter ændringer i antallet af kromosomer i de leukæmiske celler. At få mindst fem ekstra kromosomer, kaldet høj hyperdiploidi, forekommer mere almindeligt. Mindre ofte går kromosomer tabt, kaldet hypodiploidy , hvilket er forbundet med en dårligere prognose. Yderligere almindelige genetiske ændringer i B-celle ALL involverer ikke-arvelige mutationer til PAX5 og IKZF1 . I T-celle ALL, LYL1 , TAL1 , TLX1 og TLX3 kan der forekomme omlægninger.
ALLE resultater når nok af disse genetiske ændringer er til stede i en enkelt lymfoblast. I barndommen ALL findes for eksempel ofte en fusionsgen-translokation sammen med seks til otte andre ALL-relaterede genetiske ændringer. Den indledende leukæmiske lymfoblast kopierer sig selv til et for stort antal nye lymfoblaster, hvoraf ingen kan udvikle sig til fungerende lymfocytter. Disse lymfoblaster opbygges i knoglemarven og kan spredes til andre steder i kroppen, såsom lymfeknuder , mediastinum , milten , testiklerne og hjernen , hvilket fører til de almindelige symptomer på sygdommen.
Diagnose
Diagnosticering af ALL begynder med en grundig sygehistorie, fysisk undersøgelse , fuldstændig blodtælling og blodudtværinger. Mens mange symptomer på ALT kan findes ved almindelige sygdomme, giver vedvarende eller uforklarlige symptomer mistanke om kræft. Fordi mange funktioner i sygehistorien og undersøgelsen ikke er specifikke for ALLE, er det ofte nødvendigt med yderligere test. Et stort antal hvide blodlegemer og lymfoblaster i cirkulerende blod kan være mistænkelige for ALLE, fordi de indikerer en hurtig produktion af lymfoide celler i marven. Jo højere disse tal typisk peger på en dårligere prognose. Mens antallet af hvide blodlegemer ved første præsentation kan variere betydeligt, ses cirkulerende lymfoblastceller på perifert blodudtvær i de fleste tilfælde.
En knoglemarvsbiopsi giver afgørende bevis for ALLE, typisk med> 20% af alle celler, der er leukæmiske lymfoblaster. En lumbal punktering (også kendt som en rygmarven) kan afgøre, om rygsøjlen og hjernen er blevet invaderet. Involvering af hjerne og rygsøjle kan enten diagnosticeres ved bekræftelse af leukæmiske celler i lumbalpunktur eller gennem kliniske tegn på CNS -leukæmi som beskrevet ovenfor. Laboratorieundersøgelser, der kan vise abnormiteter, omfatter blodtælling, nyrefunktion, elektrolyt og leverenzymtest.
Patologisk undersøgelse, cytogenetik (især tilstedeværelsen af Philadelphia -kromosom ) og immunofenotyping fastslår, om de leukæmiske celler er myeloblastiske (neutrofiler, eosinofiler eller basofiler) eller lymfoblastiske ( B -lymfocytter eller T -lymfocytter ). Cytogenetisk test på marvprøverne kan hjælpe med at klassificere sygdom og forudsige, hvor aggressivt sygdomsforløbet vil være. Forskellige mutationer har været forbundet med kortere eller længere overlevelse. Immunhistokemisk test kan afsløre TdT- eller CALLA -antigener på overfladen af leukæmiske celler. TdT er et protein, der udtrykkes tidligt i udviklingen af pre-T og pre-B-celler, hvorimod CALLA er et antigen, der findes i 80% af ALLE tilfælde og også i "blast crisis" af CML .
Medicinsk billeddannelse (såsom ultralyd eller CT -scanning ) kan finde invasion af andre organer, sædvanligvis lunge , lever, milt, lymfeknuder, hjerne, nyrer og reproduktive organer.
akut lymfoblastisk leukæmi (ALL), et barns perifere blod, Pappenheim -plet, forstørrelse x100

knoglemarvsudstrygning (stor forstørrelse) fra en person med akut lymfoblastisk leukæmi

knoglemarvsudstrygning fra en person med akut lymfoblastisk leukæmi



Immunofenotypning
Ud over cellemorfologi og cytogenetik er immunofenotyping , en laboratorieteknik, der bruges til at identificere proteiner, der udtrykkes på deres celleoverflade, en nøglekomponent i diagnosen ALL. Den foretrukne metode til immunophenotyping er gennem flowcytometri . I de maligne lymfoblaster af ALL kan ekspression af terminal deoxynucleotidyl transferase (TdT) på celleoverfladen hjælpe med at differentiere maligne lymfocytceller fra reaktive lymfocytter , hvide blodlegemer, der reagerer normalt på en infektion i kroppen. På den anden side udtrykkes myeloperoxidase (MPO), en markør for den myeloide slægt, typisk ikke. Fordi forløber B -celle og forløber T -celler ser ens ud, kan immunofenotyping hjælpe med at differentiere undertypen af ALL og modenhedsniveauet for de ondartede hvide blodlegemer. Undertyperne af ALL som bestemt ved immunophenotype og i henhold til stadierne af modning.
| B -celle Lineage | T -celle Lineage |
|---|---|
| pre-pre-B ALL (pro-B-ALL) | forløber T- ALL |
| fælles ALT | moden T-celle ALL |
| pre-B ALLE | |
| moden B -celle ALL (Burkitt leukæmi - FAB L3) |
Et omfattende panel af monoklonale antistoffer mod celleoverflademarkører, især CD eller klynge af differentieringsmarkører, bruges til at klassificere celler efter slægt. Nedenfor er immunologiske markører forbundet med B -celle og T -celle ALL.
| Immunologiske markører | B -celle Lineage | T -celle Lineage |
|---|---|---|
| B -celle Lineage | ||
| CD19, CD22, CD79a | + | - |
| CD10 | - eller + (almindelig ALL) | |
| cytoplasmatisk Ig | - eller + (pre-B ALL) | |
| overflade Ig | - eller + (moden B-celle ALL) | |
| TdT | + | + |
| T -celle Lineage | ||
| CD2, CD3, CD4, CD5, CD7, CD8 | - | + |
| TdT | + | + |
Cytogenetik
Cytogenetisk analyse har vist forskellige proportioner og frekvenser af genetiske abnormiteter i tilfælde af ALLE fra forskellige aldersgrupper. Disse oplysninger er særligt værdifulde til klassificering og kan til dels forklare de forskellige prognoser for disse grupper. Med hensyn til genetisk analyse kan sager stratificeres i henhold til ploidi , et antal sæt kromosomer i cellen og specifikke genetiske abnormiteter, såsom translokationer . Hyperdiploide celler defineres som celler med mere end 50 kromosomer, mens hypodiploid defineres som celler med mindre end 44 kromosomer. Hyperdiploid tilfælde har en tendens til at have en god prognose, mens hypodiploid tilfælde ikke gør det. For eksempel er den mest almindelige specifikke abnormitet i B -ALL i barndommen t (12; 21) ETV6 - RUNX1 -translokation, hvor RUNX1 -genet, der koder for et protein, der er involveret i transkriptionel kontrol af hæmopoiesis , er blevet translokeret og undertrykt af ETV6 - RUNX1 fusionsprotein.
Nedenfor er en tabel med frekvenserne for nogle cytogenetiske translokationer og molekylære genetiske abnormiteter i ALLE.
| Cytogenetisk translokation | Molekylær genetisk abnormitet | % |
|---|---|---|
| kryptisk t (12; 21) | TEL - AML1 fusion | 25,4% |
| t (1; 19) (q23; p13) | E2A - PBX ( PBX1 ) fusion | 4,8% |
| t (9; 22) (q34; q11) | BCR-ABL fusion (P185) | 1,6% |
| t (4; 11) (q21; q23) | MLL - AF4 fusion | 1,6% |
| t (8; 14) (q24; q32) | IGH - MYC fusion | |
| t (11; 14) (p13; q11) | TCR - RBTN2 fusion |
Klassifikation
Fransk-amerikansk-britisk
Historisk set blev ALL inden 2008 klassificeret morfologisk ved hjælp af det fransk-amerikansk-britiske (FAB) system, der stærkt var afhængig af morfologisk vurdering. FAB-systemet tager hensyn til oplysninger om størrelse, cytoplasma , nucleoli , basofili (farve på cytoplasma) og vakuolering (boblelignende egenskaber).
| FAB -undertype | Celletype | Egenskaber | Kommentarer |
|---|---|---|---|
| ALLE - L1 | T-celle eller præ-B-celle | Små og homogene (ensartede) celler | |
| ALLE - L2 | T-celle eller præ-B-celle | Store og heterogene (varierede) celler | |
| ALLE - L3 | B -celle | Store og varierede celler med vakuoler | Moden B-celle ALL hedder også Burkitt leukæmi. Typisk dårlig prognose med standardterapi |
Mens nogle klinikere stadig bruger FAB -skemaet til at beskrive tumorcellens udseende, er meget af denne klassificering blevet opgivet på grund af dens begrænsede indvirkning på behandlingsvalg og prognostisk værdi.
Verdens Sundhedsorganisation
I 2008 blev Verdenssundhedsorganisationens klassifikation af akut lymfoblastisk leukæmi udviklet i et forsøg på at skabe et klassificeringssystem, der var mere klinisk relevant og kunne producere meningsfulde prognostiske og behandlingsbeslutninger. Dette system genkendte forskelle i genetiske, immunofenotype , molekylære og morfologiske træk fundet gennem cytogenetiske og molekylære diagnostiske tests. Denne undertyper hjælper med at bestemme prognosen og den mest passende behandling for hvert specifikt tilfælde af ALL.
WHO -undertyper relateret til ALLE er:
- B-lymfoblastisk leukæmi/lymfom
- Ikke andet angivet (NOS)
- med tilbagevendende genetiske abnormiteter
- med t (9; 22) (q34.1; q11.2); BCR-ABL1
- med t (v; 11q23.3); KMT2A omarrangeret
- med t (12; 21) (p13.2; q22.1); ETV6-RUNX1
- med t (5; 14) (q31.1; q32.3) IL3-IGH
- med t (1; 19) (q23; p13.3); TCF3-PBX1
- med hyperdiploidi
- med hypodiploidi
- T-lymfoblastisk leukæmi/lymfom
- Akutte leukæmier af tvetydig slægt
- Akut udifferentieret leukæmi
- Blandet fænotype akut leukæmi (MPAL) med t (9; 22) (q34.1; q11.2); BCR-ABL1
- MPAL med t (v; 11q23.3); KMT2A omarrangeret
- MPAL, B/myeloid, NOS
- MPAL, T/myeloid, NOS
Behandling

Formålet med behandlingen er at fremkalde en varig remission , defineret som fraværet af påviselige kræftceller i kroppen (normalt mindre end 5% blastceller i knoglemarven).
I løbet af de sidste mange årtier har der været fremskridt med at øge effektiviteten af behandlingsregimer, hvilket resulterer i øgede overlevelsesrater. Mulige behandlinger for akut leukæmi omfatter kemoterapi , steroider , strålebehandling , intensive kombinerede behandlinger (herunder knoglemarv eller stamcelletransplantationer ), målrettet terapi og/eller vækstfaktorer.
Kemoterapi
Kemoterapi er den første valg, og de fleste mennesker med ALLE modtager en kombination af medicin. Der er ingen kirurgiske muligheder på grund af den kropsdækkende fordeling af de ondartede celler . Generelt kombinerer cytotoksisk kemoterapi for ALL flere antileukæmiske lægemidler, der er skræddersyet til hver person. Kemoterapi for ALL består af tre faser: remissionsinduktion, intensivering og vedligeholdelsesbehandling.
| Fase | Beskrivelse | Agenter |
|---|---|---|
| Remission induktion | Målet er:
Skal nøje overvåge for tumorlysesyndrom efter behandlingens start Overvågning af første reaktion på behandling er vigtig, da manglende påvisning af blod eller knoglemarvsblaster inden for de første 2 uger af behandlingen har været forbundet med en højere risiko for tilbagefald
Start profylakse i CNS og administrer intratekal kemoterapi via Ommaya reservoir eller flere lumbale punkteringer |
Kombination af:
Centralnervesystemets profylakse kan opnås via:
I Philadelphia -kromosom -positive ALL kan intensiteten af indledende induktionsbehandling være mindre end traditionelt blevet givet. |
| Konsolidering/intensivering | Brug høje doser kemoterapi for yderligere at reducere tumorbyrden | Typiske protokoller bruger følgende givne blokke (varierer fra 1-3 blokke afhængig af personens risikokategori) i forskellige kombinationer af flere lægemidler:
Tilbagefald i centralnervesystemet behandles med intratekal administration af hydrocortison , methotrexat og cytarabin. |
| Vedligeholdelsesbehandling | Dræb enhver restcelle, der ikke blev dræbt ved remissionsinduktion og intensiveringsregimer
|
Typisk protokol ville omfatte:
|
På grund af tilstedeværelsen af CNS -involvering hos 10–40% af voksne med ALL ved diagnosen starter de fleste udbydere profylakse og behandling i centralnervesystemet (CNS) under induktionsfasen og fortsætter det i konsoliderings-/intensiveringsperioden.
Voksen kemoterapiregimer efterligner dem fra barndommen ALL; er imidlertid forbundet med en højere risiko for tilbagefald af sygdomme med kemoterapi alene. Det bør være kendt, at 2 undertyper af ALL (B-celle ALL og T-celle ALL) kræver særlige overvejelser, når det kommer til valg af et passende behandlingsregime hos voksne med ALL. B-celle ALL er ofte forbundet med cytogenetiske abnormiteter (specifikt t (8; 14), t (2; 8) og t (8; 22)), som kræver aggressiv terapi bestående af korte, højintensive regimer. T-celle ALL reagerer mest på cyclophosphamidholdige midler.
Da kemoterapiregimerne kan være intensive og langvarige, har mange mennesker et intravenøst kateter indsat i en stor vene (betegnet et centralt venekateter eller en Hickman -linje ) eller en Portacath , normalt placeret tæt på kravebenet, for lavere infektionsrisici og enhedens langsigtede levedygtighed. Hanner tåler normalt et længere behandlingsforløb end hunner, da testiklerne kan fungere som et reservoir for kræft.
Strålebehandling
Strålebehandling (eller strålebehandling) bruges på smertefulde knoklede områder, ved høje sygdomsbyrder eller som en del af forberedelserne til en knoglemarvstransplantation (total bestråling af kroppen). Tidligere brugte læger almindeligvis stråling i form af helhjernestråling til profylakse i centralnervesystemet for at forhindre forekomst og/eller gentagelse af leukæmi i hjernen. Nylige undersøgelser viste, at CNS -kemoterapi gav resultater som gunstige, men med færre udviklingsbivirkninger. Som et resultat heraf har brugen af helhjernestråling været mere begrænset. De fleste specialister inden for voksen leukæmi har opgivet brugen af strålebehandling til forebyggelse af CNS i stedet for at bruge intratekal kemoterapi.
Biologisk terapi
Udvælgelse af biologiske mål på grundlag af deres kombinatoriske virkninger på leukæmiske lymfoblaster kan føre til kliniske forsøg med forbedring af virkningerne af ALL behandling. Tyrosinkinasehæmmere (TKI'er), såsom imatinib , er ofte inkorporeret i behandlingsplanen for mennesker med Bcr-Abl1+ (Ph+) ALL. Denne undertype af ALL er imidlertid ofte resistent over for kombinationen af kemoterapi og TKI'er, og allogen stamcelletransplantation anbefales ofte ved tilbagefald.
Immunterapi
Kimære antigenreceptorer (CAR'er) er blevet udviklet som en lovende immunterapi for ALLE. Denne teknologi bruger et enkelt kæde variabelt fragment (scFv) designet til at genkende celleoverflademarkøren CD19 som en metode til behandling af ALL.
CD19 er et molekyle, der findes på alle B-celler og kan bruges som et middel til at skelne mellem den potentielt ondartede B-cellepopulation. I denne terapi immuniseres mus med CD19-antigenet og producerer anti-CD19-antistoffer. Hybridomer udviklet fra musemiltceller fusioneret til en myelomcellelinje kan udvikles som en kilde til cDNA, der koder for det CD19 -specifikke antistof. CDNA'et sekventeres, og sekvensen, der koder for de variable tunge og variable lette kæder af disse antistoffer, klones sammen ved hjælp af en lille peptidlinker . Denne resulterende sekvens koder for scFv. Dette kan klones ind i et transgen , der koder for det, der bliver endodomænet for CAR. Varierende arrangementer af underenheder tjener som endodomænet, men de består generelt af hængselområdet, der fæstner til scFv, en transmembranregion, den intracellulære region af et costimulatorisk molekyle, såsom CD28 , og det intracellulære domæne af CD3 -zeta, der indeholder ITAM -gentagelser . Andre sekvenser, der ofte er inkluderet, er: 4-1bb og OX40 . Den endelige transgensekvens, der indeholder scFv- og endodomainsekvenserne, indsættes derefter i immuneffektorceller, der opnås fra personen og ekspanderes in vitro . I forsøg har disse været en type T-celle, der er i stand til cytotoksicitet .
Indsættelse af DNA'et i effektorcellen kan udføres ved flere metoder. Normalt gøres dette ved hjælp af et lentivirus, der koder for transgenet. Pseudotypede, selvinaktiverende lentivirus er en effektiv metode til stabil indsættelse af et ønsket transgen i målcellen. Andre metoder omfatter elektroporation og transfektion , men disse er begrænsede i deres effektivitet, da transgenekspression falder over tid.
De genmodificerede effektorceller transplanteres derefter tilbage i personen. Typisk udføres denne proces i forbindelse med et konditioneringsregime, såsom cyclophosphamid , som har vist sig at forstærke virkningerne af infunderede T-celler. Denne effekt er blevet tilskrevet at lave et immunologisk rum, inden for hvilket cellerne befolker sig. Processen som helhed resulterer i en effektorcelle , typisk en T-celle, der kan genkende et tumorcelle- antigen på en måde, der er uafhængig af det store histokompatibilitetskompleks, og som kan starte en cytotoksisk reaktion.
I 2017 blev tisagenlecleucel godkendt af FDA som en CAR-T- behandling for mennesker med akut B-celle lymfoblastisk leukæmi, som ikke reagerede tilstrækkeligt på andre behandlinger eller er faldet tilbage. I en 22-dages proces tilpasses "stoffet" til hver person. T -celler renset fra hver person modificeres af en virus, der indsætter gener, der koder for en kimær antigenreceptor i deres DNA, en der genkender leukæmiceller.
Tilbagefald ALLE
Typisk har mennesker, der oplever et tilbagefald i deres ALL efter indledende behandling, en dårligere prognose end dem, der forbliver i fuldstændig remission efter induktionsterapi. Det er usandsynligt, at tilbagevendende leukæmi vil reagere positivt på det standard kemoterapiregime, der oprindeligt blev implementeret, og i stedet bør disse mennesker prøves på genindledningskemoterapi efterfulgt af allogen knoglemarvstransplantation . Disse mennesker i tilbagefald kan også modtage blinatumomab , da det har vist sig at øge remissionsrater og generelle overlevelsesrater uden øgede toksiske virkninger.
Lav dosis palliativ stråling kan også bidrage til at reducere tumorbyrden i eller uden for centralnervesystemet og lindre nogle symptomer.
For nylig har der også været bevis og godkendelse af brug af dasatinib , en tyrosinkinasehæmmer. Det har vist effekt i tilfælde af mennesker med Ph1-positive og imatinib- resistente ALL, men der skal foretages mere forskning om langsigtet overlevelse og tid til tilbagefald.
Bivirkninger
Kemoterapier eller stamcelletransplantationer kan kræve en trombocyttransfusion for at forhindre blødning. Desuden kan patienter, der gennemgår en stamcelletransplantation, udvikle en transplantat-versus-host-sygdom (GvHD). Det blev evalueret, om mesenkymale stromaceller kan bruges til at forhindre en GvHD. Beviserne er meget usikre på den terapeutiske virkning af mesenkymale stromaceller til behandling af transplantat-versus-host-sygdomme efter en stamcelletransplantation på alle årsager til dødelighed og fuldstændig forsvinden af kroniske akutte transplantat-versus-host sygdomme. Mesenkymale stromaceller kan resultere i ringe eller ingen forskel i dødeligheden af alle årsager, tilbagefald af malign sygdom og forekomst af akutte og kroniske transplantat-versus-host-sygdomme, hvis de bruges af profylaktisk årsag.
Støttende terapi
Tilføjelse af fysiske øvelser til standardbehandlingen for voksne patienter med hæmatologiske sygdomme som ALL kan resultere i lille eller ingen forskel i dødelighed, livskvalitet og fysisk funktion. Disse øvelser kan resultere i en lille reduktion i depression. Desuden reducerer aerobe fysiske øvelser sandsynligvis træthed. Beviserne er meget usikre på virkningen på angst og alvorlige bivirkninger.
Genterapi
Brexucabtagene autoleucel (Tecartus) blev godkendt til behandling af voksne med tilbagefald eller ildfast B-celleforløber akut lymfoblastisk leukæmi i oktober 2021.
Hver dosis brexucabtagene autoleucel er en tilpasset behandling skabt ved hjælp af modtagerens eget immunsystem for at hjælpe med at bekæmpe lymfom. Modtagerens T -celler , en type hvide blodlegemer, indsamles og genetisk modificeres til at omfatte et nyt gen, der letter målretning og aflivning af lymfomceller. Disse modificerede T -celler infunderes derefter tilbage i modtageren.
Prognose
Før udviklingen af kemoterapiregimer og hæmatopoietisk stamcelletransplantation overlevede børn en median længde på 3 måneder, hovedsageligt på grund af enten infektion eller blødning. Siden fremkomsten af kemoterapi er prognosen for leukæmi hos børn blevet stærkt forbedret, og børn med ALL skønnes at have en 95% sandsynlighed for at opnå en vellykket remission efter 4 ugers behandlingsstart. Mennesker i pædiatrisk pleje med ALL i udviklede lande har en overlevelsesrate på mere end 80% over fem år. Det anslås, at 60–80% af voksne, der gennemgår induktionskemoterapi, opnår fuldstændig remission efter 4 uger, og dem over 70 år har en kur på 5%. Hutter JJ (juni 2010). "Barndomsleukæmi". Pædiatri i gennemgang . 31 (6): 234–41. doi : 10.1542/pir.31-6-234 . PMID 20516235 .</ref>

Imidlertid er der forskellige prognoser for ALLE blandt individer afhængigt af en række faktorer:
- Køn: Kvinder har en tendens til at klare sig bedre end mænd.
- Etnicitet: Kaukasiere er mere tilbøjelige til at udvikle akut leukæmi end afroamerikanere , asiater eller latinamerikanere . Men de har også en tendens til at have en bedre prognose end ikke-kaukasiere.
- Alder ved diagnosen: børn i alderen 1-10 år er mest tilbøjelige til at udvikle ALT og blive helbredt for det. Tilfælde hos ældre mennesker er mere tilbøjelige til at skyldes kromosomale abnormiteter (f.eks. Philadelphia -kromosomet), der gør behandlingen vanskeligere og prognoserne dårligere. Ældre mennesker har sandsynligvis også co-morbide medicinske tilstande, der gør det endnu vanskeligere at tolerere ALT behandling.
- Antallet af hvide blodlegemer ved diagnosen større end 30.000 (B-ALL) eller 100.000 (T-ALL) er forbundet med dårligere resultater
- Kræft, der spreder sig til centralnervesystemet ( hjerne eller rygmarv ) har dårligere resultater.
- Morfologiske, immunologiske og genetiske undertyper
- Personens reaktion på indledende behandling og længere tid krævet (mere end 4 uger) for at nå fuldstændig remission
- Tidligt tilbagefald af ALLE
- Minimal restsygdom
- Genetiske lidelser , såsom Downs syndrom og andre kromosomale abnormiteter (aneuoploidi og translokationer)
| Faktor | Ufordelagtig | Favorit |
|---|---|---|
| Alder | <2 eller> 10 år | 3-5 år |
| Køn | Han | Kvinde |
| Race | Sort | Kaukasisk |
| Organomegali | Til stede | Fraværende |
| Mediastinal masse | Til stede | Fraværende |
| CVS involvering | Til stede | Fraværende |
| Leukocytantal | B-ALL> 30.000 mm 3 T-ALL> 100.000 mm 3 | Lav |
| Hæmogblobinkoncentration | > 10 g/dl | <10 g/dl |
| Celletype | Ikke lymfatisk | Lymfoid |
| Celleafstamning | Pre B -celle +
T-ALL (børn) |
Tidlig Pre B -celle |
| Karyotype | Translokation | Hyperdiploidi |
| Svar på behandling | Langsom
> 1 uge for at fjerne blaster fra blod |
Hurtig
<1 uge for at fjerne blaster fra blod |
| Tid til eftergivelse | > 4 uger | <4 uger |
| Minimal restsygdom | Positiv efter 3-6 måneder | Negativ ved 1 måned (børn) eller 3 måneder (voksne) |
Cytogenetik, studiet af karakteristiske store ændringer i kromosomerne i kræftceller , er en vigtig forudsigelse for udfaldet. Nogle cytogenetiske undertyper har en dårligere prognose end andre. Disse omfatter:
- Personer med t (9,22) positive-ALL (30% af voksne ALLE tilfælde) og andre Bcr-abl- omlejrede leukæmier har større sandsynlighed for en dårlig prognose, men overlevelsesraten kan stige ved behandling bestående af kemoterapi og Bcr-abl tyrosinkinasehæmmere.
- En translokation mellem kromosomer 4 og 11 forekommer i cirka 4% af tilfældene og er mest almindelig hos spædbørn under 12 måneder.
| Cytogenetisk ændring | Risikokategori |
|---|---|
| Philadelphia kromosom | Dårlig prognose |
| t (4; 11) (q21; q23) | Dårlig prognose |
| t (8; 14) (q24.1; q32) | Dårlig prognose |
| Kompleks karyotype (mere end fire abnormiteter) | Dårlig prognose |
| Lav hypodiploidi eller nær triploidi | Dårlig prognose |
| Sletning af kromosom 7 | Dårlig prognose |
| Trisomi 8 | Dårlig prognose |
| Høj hyperdiploidi (trisomi 4, 10, 17) | God prognose |
| del (9p) | God prognose |
- Hyperdiploidy (> 50 kromosomer) og t (12; 21) er gode prognostiske faktorer og udgør også 50% af pædiatriske ALLE tilfælde.
| Prognose | Cytogenetiske fund |
|---|---|
| Favorit | Hyperdiploidy> 50; t (12; 21) |
| Mellemliggende | Hyperdiploidy 47–50; Normal (diploid); del (6q); Omlægninger af 8q24 |
| Ufordelagtig | Hypodiploidy-nær haploidi; Nær tetraploidi; del (17p); t (9; 22); t (11q23) |
Uklassificeret ALL anses for at have en mellemliggende prognoserisiko, et sted mellem de gode og dårlige risikokategorier.
Epidemiologi
ALLE ramte omkring 876.000 mennesker og resulterede i 111.000 dødsfald globalt i 2015. Det forekommer hos både børn og voksne med de højeste satser set mellem tre og syv år. Omkring 75% af tilfældene opstår før 6 -årsalderen med en sekundær stigning efter 40 -årsalderen. Det anslås at påvirke 1 ud af 1500 børn.
Med hensyn til de brede aldersprofiler for de berørte forekommer ALLE nyligt hos omkring 1,7 pr. 100.000 mennesker om året. ALL repræsenterer cirka 20% af voksne og 80% af barndomsleukæmier, hvilket gør det til den mest almindelige kræft hos børn. Selvom 80 til 90% af børnene vil have et langsigtet fuldstændigt svar med behandlingen, er det fortsat den førende årsag til kræftrelaterede dødsfald blandt børn. 85% af tilfældene er af B-celle-slægt og har lige mange tilfælde hos både mænd og kvinder. De resterende 15% af T-cellelinjen har en mandlig overvægt.
Globalt forekommer ALLE typisk oftere hos kaukasiere, latinamerikanere og latinamerikanere end hos afrikanere. I USA er ALLE mere almindelige hos børn fra kaukasisk (36 tilfælde/millioner) og spansktalende (41 tilfælde/millioner) herkomst sammenlignet med børn fra afrikansk (15 tilfælde/million) afstamning.
Graviditet
Leukæmi er sjældent forbundet med graviditet, og rammer kun ca. 1 ud af 10.000 gravide. Behandlingen af leukæmi hos en gravid person afhænger primært af typen af leukæmi. Akutte leukæmier kræver normalt hurtig, aggressiv behandling på trods af betydelige risici for graviditetstab og fosterskader , især hvis kemoterapi gives i den udviklingsfølsomme første trimester .
Referencer
eksterne links
- Akut lymfatisk leukæmi hos American Cancer Society
- Barndom ALL behandling på National Cancer Institute
| Klassifikation | |
|---|---|
| Eksterne ressourcer |