
Genotype -første tilgang - Genotype-first approach
Den genotype -første tilgang er en type strategi, der bruges i genetiske epidemiologiske undersøgelser til at knytte specifikke genotyper til tilsyneladende kliniske fænotyper af en kompleks sygdom eller egenskab. I modsætning til "fænotype-først", den traditionelle strategi, der hidtil har været styrende for genom-dækkende associeringsstudier (GWAS), karakteriserer denne tilgang individer først ved en statistisk almindelig genotype baseret på molekylære test forud for klinisk fænotypisk klassificering. Denne grupperingsmetode fører til patientevalueringer baseret på en delt genetisk ætiologi for de observerede fænotyper, uanset deres mistanke om diagnose. Således kan denne tilgang forhindre indledende fænotypisk bias og muliggøre identifikation af gener, der udgør et væsentligt bidrag til sygdomens ætiologi .
Denne fremgangsmåde er upåvirket af fænotypisk heterogenitet, ufuldstændig penetrans og ekspressivitetsniveauer. Derfor er det nyttigt i komplekse sygdomme, der også overlapper hinanden, såsom autismespektrumforstyrrelse og intellektuel funktionsnedsættelse , hvilket gør det muligt at skelne mellem sygdommene og specifikke undertyper af sygdommen baseret på det genomiske indhold, der kan bestemmes.
I øjeblikket bruges genotype-første tilgang primært til forskningsmål. Implikationerne fra disse undersøgelser kan imidlertid have værdifulde kliniske anvendelser, herunder forbedret diagnose, rådgivning og støttegrupper for personer med samme genetiske ætiologi.
Baggrund
I første omgang blev ideen om at identificere genotypen for individer og efterfølgende deres tilknyttede fænotype (r) først brugt i tidlige cytogenetiske undersøgelser. Omkring 1960 førte opdagelsen af Trisomy 21 til erkendelsen af, at genetik kunne bruges til at forudsige fænotype (r). Fra 1960'erne til 1990'erne blev cytogenetiske teknikker såsom kromosombinding og fluorescens in situ hybridisering (FISH) brugt til at identificere og fænotypisk karakterisere patienter med kromosomale abnormiteter.
Komplekse sygdomme og egenskaber udgør mange vanskeligheder ved epidemiologiske undersøgelser på grund af deres art som multifaktorielle sygdomme. Mere end ét gen kan ligge til grund for en kompleks sygdom og bidrager generelt med en mindre effekt end hvad der observeres ved monogene sygdomme ( Mendelske sygdomme ). Desuden udviser mange af disse komplekse sygdomme forskellige fænotyper samt en bred vifte af udtryksfuldhed og gennemtrængelighed. Gener kan også være pleiotropiske og tegner sig for mange tilsyneladende forskellige kliniske fænotyper. Disse egenskaber begrænser både forskning og kliniske undersøgelses evne til at udpege kausale gener eller varianter til de observerede fænotyper og til at klassificere lidelser.
Klinikere begynder at erkende behovet for at klassificere genomiske sygdomme efter en fælles genotype frem for en fælles fænotype, og hvordan genotype-første tilgang kan gavne dette formål.
Metoder
Flere metoder kan bruges med en genotype-første tilgang, men følgende trin er normalt inkluderet:
- Etablering af en undersøgelsespopulation og genotypning
- Analyse af genomiske varianter af interesse fundet i undersøgelsespopulationen
- Studiepopulationer samles baseret på genotype
- Forening af genotype til fænotype (r) inden for respektive gruppe
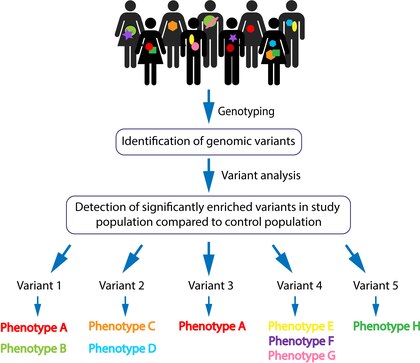
Genotypebestemmelsen genereres ved hjælp af næste generations sekventeringsteknologier (herunder helgenomsekvensering og eksomsekventering ) og mikroarrayanalyser . Rådataene analyseres derefter statistisk for populationsbaseret frekvens af varianterne. Almindelige varianter filtreres fra, og patogenicitet bestemmes selvom forudsagte genetiske implikationer. Disse trin giver mulighed for identifikation af formodede stærkt penetrante varianter og deres specifikke locus . De udvalgte varianter resekventeres normalt til validering (ved målrettet Sanger -sekventering ). Validerede genomiske varianter kan derefter analyseres for tilbagefald blandt berørte individer i kohorten. Patogenicitet af en genomisk variant er statistisk baseret på dens signifikant rigelige tilstedeværelse i de berørte i forhold til de upåvirkede individer, ikke udelukkende på variantens skadelighed. En kandidatvariant kan derefter associeres med en delt fænotype med ambitionen om, at efterhånden som flere patienter, der bar den samme variant med den samme fænotype, vil blive identificeret, kan der skabes en stærkere association. Endelig afgrænses der mellem en specifik variant til associerede kliniske fænotyper [Figur 1].
Kliniske implikationer og eksempler
Den genotype-første tilgang er blevet brugt til at diagnosticere patienter med sjældne sygdomme, identificere nye sygdomsgenotype-fænotyperforeninger og karakterisere usædvanlige eller heterogene sygdomme baseret på patientens genotype. I 2014 blev den første genotype-metode anvendt til at vurdere sjældne og lavfrekvente varianter i den finske befolkning. Da den finske befolkning er isoleret og for nylig har gennemgået en befolkningsflaskehals i forhold til andre lande, giver den to hovedfordele ved genotype-første undersøgelser. Skadelige varianter findes ved højere frekvenser inden for et mindre spektrum af sjældne varianter i flaskehalsede grundlæggerpopulationer. Ved at sammenligne de varianter, der blev fundet ved hjælp af hele-exome-sekventering (WES) i den finske befolkning med WES fra en kontrolgruppe af ikke-finske europæere, blev varighedstab (LOF) -varianter set med en højere frekvens i den finske befolkning. Fænotyperne for finske individer med disse LOF -varianter blev derefter analyseret for at fastslå nye genotype -fænotypeforeninger. Disse påviste foreninger inkluderede en, der kunne være embryonal dødelig, oplysninger, der muligvis ikke var blevet opdaget i forskning ved hjælp af en fænotype-første tilgang. Derudover opdagede forskere også nye splejsningsvarianter i LPA -genet, der reducerer apolipoprotein A -niveauer og tilbyder en beskyttende fænotype mod hjerte -kar -sygdomme.
Genotype-første vurdering er ved at blive standardmetoden for klinisk diagnose af komplekse heterogene sygdomme. Mikroduplikations- og mikrodeletionssyndromer har en række karakteristika, herunder intellektuel funktionsnedsættelse og udviklingsforsinkelse , som varierer i sværhedsgrad, hvilket gør patienter med disse syndromer meget vanskelige at diagnosticere. Siden udviklingen af næste generations sekventeringsteknologier har klinikere været i stand til at bruge en genotype-første tilgang til at gruppere disse patienter baseret på deres mikrosletning eller kopiering og dokumentere sygdomsegenskaberne i disse grupper. Kromosomal mikroarrayanalyse bruges især klinisk til at hjælpe med at diagnosticere patienter med mikrodeletion og mikrodulplikationssyndromer. Ved sygdomme, såsom Autismespektrumforstyrrelse (ASD), hvor differentiering af patienter i sygdomsundertypegrupper baseret på fænotype er udfordrende, tillader genotype-første undersøgelser klassificering af patienter i undertyper baseret på deres genetik. Dette vil igen give en større forståelse af de genetiske årsager til ASD og kunne i fremtiden definere specifikke undertyper af ASD for patienter, der skal diagnosticeres med.
Genotype-første forskning, gennem identifikation af nye sygdomsassocierede gener, kan også gavne farmaceutiske virksomheder og lægemiddeludvikling. For komplekse sygdomme ved hjælp af fænotype første genforening er udvikling af terapier ofte uden succes på grund af flere gener, der bidrager til en sygdom. Med genotype-første associationer identificeres det potentielle terapeutiske mål først.
Fordele og begrænsninger
Fordele
- Et skift til at karakterisere individer efter en fælles genotype frem for den kliniske præsentation vil muliggøre klassificering af nye syndromer og den genetiske klassificering af en bestemt sygdomssubtyper, idet sekventering bliver billigere, hurtigere og mere effektiv.
- Arv af en genomisk variant fra en sund forælder ville ikke resultere i dens udelukkelse fra variantanalyse og derved redegøre for modifikatorernes rolle for fænotypisk resultat.
- Denne fremgangsmåde er upåvirket af fænotypisk heterogenitet, ufuldstændig penetration og udtryksfuldhed.
- Denne tilgang bidrager til at studere både ekspressive, pleiotropi og sporadiske mutationer.
- Denne tilgang undersøger stærkt penetrante mutationer, der er forbundet med sygdommen uanset den genetiske baggrund.
- Omfattende og detaljeret fænotypering er mulig, selv med et lille antal patienter med fælles genetisk ætiologi.
- Denne tilgang kan identificere atypiske præsentationer af sygdom, når de bruges diagnostisk.
Begrænsninger
- Fænotypen kan ændre sig over tid (f.eks. Bliver mere alvorlig, ændring i fysisk placering), hvilket gør genotype-første undersøgelser til en antagelse om variantens rolle i sygdomsmanifestation på et bestemt tidspunkt. Derfor er opfølgning i længderetning vigtig, for at genotype -fænotype -foreningen kan værdiansættes med tiden og undersøge sygdommens prognose.
- Identificerede varianter, der kan bidrage til en mild fænotype eller til en række fænotyper, ville ikke være gavnlige ved bestemmelse af diagnose og prognose. Men i fremtiden, da flere sygdomsundertyper er klassificeret, kan milde fænotyper have mere relevans.
- Genotype-fænotypeforening er afhængig af præsentationen af klinisk genkendelige fænotyper.
- Som det ses i andre genomforeningsstudier, kan denne fremgangsmåde generere varianter af ukendt betydning , især når de bruges diagnostisk.
Se også
- Kompleks sygdom
- Genome-wide association study (GWAS)
- Microarray
- Hele eksome sekventering (WES)
- Hele genom-sekventering (WGS)