
Liposarcoma - Liposarcoma
| Liposarkom | |
|---|---|
 | |
| Histopatologi af liposarkom, H & E -plet: - | |
| Specialitet | Dermatologi , generel kirurgi onkologi |
| Symptomer | Klump under huden, smerter, hævelse, organ dysfunktion |
Liposarkomer er den mest almindelige undertype af blødt væv sarkomer , tegner sig for mindst 20% af alle sarkomer hos voksne. Bløddelssarkomer er sjældne neoplasmer med over 150 forskellige histologiske undertyper eller former. Sarkomer, som udgør ~ 1% af alle voksne maligniteter, er ondartede tumorer, der udvikler sig fra stamcellerne i mesenkymale (dvs. bindevæv) væv såsom: osteosarkomer, der opstår fra osteoprogenitorcellerne (dvs. forløber) celler i de modne osteocytter i knogler væv; de fibrosarkomer, der opstår fra fibrocytternes forstadieceller i bindevæv ; og rabdomyosarkomerne, der stammer fra myocytternes forstadieceller i muskelvæv . Liposarkomer opstår fra precursor lipoblasts af adipocytter (dvs. fedtceller) i adipose (dvs. fedt) væv . Fedtvæv er fordelt i hele kroppen, herunder steder som de dybe og mere overfladiske lag af subkutane væv samt på mindre kirurgisk tilgængelige steder som retroperitoneum (dvs. plads bag bughulen ) og visceralt fedt inde i bughulen .
Alle liposarkomer består af mindst nogle celler, der ligner fedtceller, når de undersøges for deres histopatologiske udseende under et mikroskop. Liposarkomerne har imidlertid flere former baseret på forskelle i deres kliniske præsentationer (f.eks. Alder, kønspræferencer, tumorsider, tegn og symptomer ), sværhedsgrader (dvs. potentiale for at invadere lokale væv, gentage sig efter kirurgisk fjernelse og metastasere til distale væv), genetiske abnormiteter , prognoser og foretrukne behandlingsregimer. The World Health Organization i 2020 omklassificeret liposarkomer i fem mere eller mindre forskellige former: 1) atypiske lipomatøs tumor / veldifferentieret liposarkom; 2) dedifferentieret liposarkom; 3) myxoid liposarkom; 4) pleomorf liposarcom; og 5) myxoid pleomorf liposarkom. ( Pleomorf angiver tilstedeværelsen af celler, der har unormale og ofte store variationer i deres størrelse og form og/eller størrelsen og formen på deres kerner.)
Selvom liposarcoma-former er klassificeret som aggressiv og ondartet, eller i tilfælde af atypisk lipomatøs tumor/veldifferentieret liposarcoma som relativt ikke-aggressive og godartede, kan alle fem liposarcomformer infiltrere lokalt for at skade nærliggende væv og organer, forekomme i kirurgisk utilgængelige steder ved siden af vitale organer (f.eks. retroperitoneum), gentages efter kirurgisk fjernelse og fremskridt til livstruende sygdomme. Undersøgelser har fundet ud af, at alle fem liposarkomformer, mens de normalt kan behandles i det mindste i første omgang ved kirurgisk resektion, ofte kun er marginalt lydhøre over for i øjeblikket anvendte kemoterapi- og strålebehandlinger . Liposarkomerne kræver en lang række yderligere undersøgelser for at bestemme deres lydhørhed over for forskellige strålebehandling , kemoterapi og mere nye behandlingsregimer, der bruges individuelt og i forskellige kombinationer, der, hvor det er muligt, vil omfatte kirurgisk fjernelse.
Former af liposarkomer
Liposarkomer er generelt store tumorer (> 10 cm), men kan være af næsten enhver størrelse. De forekommer hovedsageligt hos voksne, og kun 0,7% af tilfældene forekommer hos dem <16 år. Hos voksne forekommer liposarkomer overvejende i og efter middelalderen. De meget sjældne tilfælde, der forekommer hos børn og unge, diagnosticeres overvejende som myxoid liposarkom.
De fem liposarkomformer skal ikke kun skelnes fra hinanden, men også fra visse andre bløde vævstumorer. Disse andre tumorer sammen med nogle af deres kendetegnende histopatologiske egenskaber er: 1) dysplastiske lipomer (dvs. godartede humorer, der har steder med vævsnekrose og neoplastiske fedtceller med variabel størrelse indeholdende kerner af variabel størrelse/form ; disse neoplastiske celler, i modsætning til de fleste neoplastiske celler i liposarkomer, ikke overudtrvkker den MDM2 genet); 2) atypiske spindelcellelipomer (dvs. godartede tumorer med let atypiske spindelformede celler i et fibrøst-til-myxoid stroma blandet med vakuolerede lipoblaster og adipocytter i variabel størrelse med atypiske kerner; 3) pleomorfe lipomer (dvs. godartede tumorer karakteriseret ved kæmpeceller med overlappende kerner); og 4) solitære fibrøse tumorer (dvs. tumorer, hvoraf op til 22% udviser malign adfærd, der består af spindel- eller ægformede celler i en kollagen baggrundstroma blandet med blodkar med en karakteristisk staghornform).
Atypisk lipomatøs tumor/veldifferentieret liposarkom
Tilsammen udgør atypiske lipomatiske tumorer (ALAT'er) og veldifferentierede liposarkomer (WDL'er) 40% til 45% af alle liposarkomer. De metastaserer sjældent og nogensinde og betragtes derfor som godartede eller premaligne tumorer. De er imidlertid lokalt invasive og kan omdannes til et mere aggressivt og potentielt metastaserende liposarkom, dvs. et dedifferentieret liposarkom. Endvidere kan en kirurgisk fjernet atypisk lipomatøs tumor/veldifferentieret liposarkom gentage sig som et dedifferentieret liposarkom.
Præsentation

ALAT'er og WDL'er betragtes som praktisk talt identiske tumorer, bortset fra at ALT'er pr. Definition betegner tumorer, der udvikler sig i arme eller ben, mens WDL'er betegner tumorer, der udvikler sig på mindre kirurgisk tilgængelige steder, såsom de dybe, centralt placerede bløde væv i retroperitoneum , paratesticulær region ( dvs. område inden i pungen, herunder testiklerne , sædstrengen , tunika i testiklerne , epididymis og blindtarmens blindtarm ), mundhule og øjenhule . Denne terminologi har prognostiske konsekvenser: mindre end 7% af ALAT -tumorer konverterer til dedifferentierede liposarkomer inden for en mediantid på 7 år, mens 17% af WDL -tumorer konverterer til dette mere ondartede liposarkom inden for en mediantid på 8 år. ALT og WDL (i det følgende kaldet ALT/WDL) tumorer findes typisk i midaldrende og ældre individer som langsomt forstørrende masser, der har en tendens til at være større og på et mere avanceret stadium, når de befinder sig i dybe væv. Disse tumorer er sædvanligvis ikke smertefulde, og hvis de er placeret overfladisk, let synlige; de kan også forårsage omfattende ødem (dvs. hævelse på grund af lokal væskeansamling) i involverede områder som låret (se tilstødende figur) på grund af deres invasion i blodet og/eller lymfekar, der dræner tumorens sted. Dybtliggende ALT/WDL-tumorer kan være asymptomatiske, men afhængigt af deres placering kan de forårsage alvorlige tegn og/eller symptomer på funktionsfejl i et af de forskellige organer, de infiltrerer. Disse organer indbefatter organer tæt på eller i retroperitoneum (f.eks. Tarm, nyre og nyrens urinleder ); den paratesticular region; mediastinum (f.eks. luftrør og lunges store bronkier ); og hovedet (f.eks. retrobulbarrummet bag øjet globus).
Patologi

Histopatologisk er ALT/WDL-tumorer opdelt i adipocytiske/lipomlignende, skleroserende og inflammatoriske varianter, hvor adipocyt/lipoma-lignende er den mest almindelige. Adipocytiske/lipomlignende ALT/WDL-tumorer består af lobuler af modne fedtceller, der er krydset variabelt med uregelmæssig fibrøs septa (se det tilstødende H&E farvede mikrofotografi ). Scleroserende ALT/WDL -tumorer, den anden mest almindelige variant, udvikler sig primært i retroperitoneale og paratesticulære områder; den består af spredte, atypiske stromaceller inden for en kollagen (dvs. kollagenholdig ) stromal vævsbaggrund . Sjældne vacuole holdigt lipoblasts befolke dette væv. Inflammatoriske ALT/WDL -tumorer er den sjældneste variant. de forekommer hyppigst i retroperitoneum og består af kroniske inflammatoriske celler, f.eks. lymfocytter og plasmaceller plus lejlighedsvise lymfeknude-lignende follikler spredt gennem en vævsbaggrund, der indeholder fedtceller .
Genetik
De neoplastiske celler i ALT/WDL-tumorer indeholder et eller flere ekstra ringformede små supernumerære markørkromosomer (sSMC) eller et unormalt kæmpemarkørkromosom (dvs. et tidligere normalt kromosom, der gøres unormalt ved at have en kopiering af dele af sit eget eller et eller flere andre kromosomers genetiske materiale). Disse unormale kromosomer indeholder ekstra kopier af kromosom 12 's lange arm (også kaldet q-armen ) ved bånd 13 til 15. Denne strækning af kromosom 12 inkluderer MDM2 proto-onkogen (et potentielt tumorfremkaldende gen, når det overudtrykkes ) placeret ved båndet 15 og CDK4 (et gen, der ved overudtryk fremmer udviklingen af forskellige tumorer) placeret ved bånd 14.1. Den amplificiation (dvs. øget kopier af et gen uden en proportional stigning i andre gener) af disse to gener er en meget følsom og specifik indikator for, at en liposarkom er enten en ALT / WDL eller en dedifferentierede liposarkom snarere end nogen anden liposarkom eller lipoma formular. Ud over MDM2- og CDK4 -generne indeholder dette bånd 13-15 kromosomområde også TSPAN31- og HMGA2 -generne , som, når de overudtrykkes, er forbundet med forskellige tumorer og/eller kræftformer. En eller flere af disse overudtrykte gener, det er blevet foreslået, fremmer og/eller bidrager til udvikling og/eller progression af ALT/WDL -tumorer.
Diagnose
Diagnosen ALT/WDL -tumorer stilles baseret på funktionerne i deres kliniske præsentationer, histopatologi og genetiske fund. Især påvisning i ALT/WDL-tumorcellerne af et overudtrykt MDM2- eller CDK4- gen eller tilstedeværelsen af enten det specifikke ALT/WDL-associerede sSMC eller kæmpemarkørkromosom (som defineret ved næste generations DNA-sekventering , komparativ genomisk hybridisering og/ eller højt specialiserede cytogenetiske G -bandingsanalyser ) understøtter kraftigt diagnosen ALT/WDL eller dedifferentieret liposarkom. Den kliniske præsentation og histopatologiske forskelle mellem de to sidstnævnte liposarkomformer hjælper normalt med at skelne mellem dem.
Behandling og prognose
ALT/WDL tumorer behandles ved radikal kirurgisk resektion for at fjerne alt tumor neoplastisk væv. Disse tumorer gentager sig imidlertid lokalt i 30-50% af tilfældene. Tilbagefald forekommer oftest i tumorer placeret på mindre tilgængelige steder, såsom dem i retroperitoneum, mediastinum og sædstreng. Disse mindre kirurgisk vurderbare tumorer har tendens til at gentage sig gentagne gange og kan i sidste ende forårsage død på grund af deres skadelige virkninger på vitale organer. Selvom ALT/WDL -tumorer har meget lidt potentiale til metastasering , vil omkring 10% konvertere til en åbenlyst ondartet og potentielt metastaserende liposarkomform, dedifferentieret liposarkom. Mediantiden for denne ondartede transformation er omkring 7-9 år. Derudover kan et kirurgisk fjernet ALT/WDL gentage sig efter et variabelt interval som et dedifferentieret liposarkom. Et stort randomiseret kontrolleret forsøg, der sammenlignede strålebehandling efterfulgt af kirurgi med kirurgi alene i ALT/WDL -tumorer, fandt lille forskel mellem de to regimer. Mindre undersøgelser med selektive hæmmere af proteinprodukterne fra CDK4- eller MDM2 -generne impliceret i ALT/WDL har i bedste fald kun vist beskedne virkninger. Yderligere undersøgelser af disse eller helt nye behandlingsregimer er under undersøgelse. En gennemgangsundersøgelse i 2012 rapporterede, at 5 og 10 års overlevelsesrater for personer med ALAT/WDL var henholdsvis 100% og 87%.
Nye terapier
De nye terapier ved ALT/WDL er de samme som dem, der er angivet i afsnittet Ny terapi af Dedifferentieret liposarkom.
Dedifferentieret liposarkom
Dedifferentierede liposarkomer er ondartede tumorer, som i ~ 10% af tilfældene udvikler sig i en eksisterende atypisk lipomatøs tumor/veldifferentieret liposarcoma (ALT/WDL) tumor eller på stedet, hvor en ALT/WPL-tumor blev fjernet kirurgisk. Personer med en de novo -diagnose af denne tumor kan have haft en ALT/WDL, der udviklede sig til et dedifferentieret liposarkom, men gik opdaget, fordi det udviklede sig asymptomatisk på et meget sekvestreret sted, såsom retroperitoneum eller bughulen. Mange af de dedifferentierede liposarcoma -tumors kliniske og genetiske træk ligner dem, der findes i ALT/WDL -tumorer.
Præsentation
Dedifferentierede lipoosarkomer (DDL) forekommer hyppigst hos midaldrende og ældre voksne med en maksimal forekomst i deres sjette til ottende årti. Sjældent har disse tumorer udviklet sig hos børn og unge. DDL -tumorer forekommer oftest i retroperitonealrummet, men ligner ALT/WDL kan forekomme i ekstremiteterne, paratestikulært område, mediastinum, hoved eller hals. Mindre end 1% af alle DDL'er udvikler sig i overfladiske bløde væv eller i øjenhulen. Ved præsentation er DDL-tumorer typisk smertefri, store, kan have været langsomt og gradvist forstørret i årevis, og på rutinemæssige røntgenstråler indeholder områder med calciumaflejring (eksemplificeret ved fig. 1 i afsnittet Histopathology of liposarcomas). Mindre almindeligt har ramte personer tegn og/eller symptomer på grund af deres tumors påvirkning af et organ (f.eks. Mavesmerter forårsaget af blokering af tarmene eller urinvejsobstruktion forårsaget af blokering af urinrøret ). Meget sjældent har personer med DDL et eller flere tegn eller symptomer på kronisk betændelse (se B-symptomer ) og/eller et af de endokrine , neurologiske , mucokutane , hæmatologiske eller andre vævsrelaterede paraneoplastiske syndromer . Tegn og symptomer på kronisk inflammation og de forskellige paraneoplastiske syndromer er forårsaget af tumorenes udskillelse af cytokiner , hormoner , prostaglandiner og/eller andre systemisk virkende midler; de forsvinder fuldstændigt, efter at DDL er behandlet med succes.
Patologi
Det histopatologiske udseende af DDL -tumorer (se fig. 2 i nedenstående histopatologi for liposarkomer) varierer meget, men udviser oftest træk ved udifferentierede pleomorfe sarkomer (som er tumorer tætbefolket med celler med variabel størrelse og form, der indeholder variabelt størrelse og formede kerner ) eller spindelcellesarkomer (som er tumorer bestående af spindelformede celler i bindevævsbaggrund ). Forskellige dele af DDL tumorer viser ofte variationer i optrædener af deres baggrund bindevæv: disse væv kan være myxoid (dvs. bestående af en klar, slim -lignende stof, som ved farvning under anvendelse af en standard H & E stain metoden synes mere blå eller lilla end den røde farve af normale væv) eller myxocollagenous (dvs. stor kollagen fiberindhold i en myxoid baggrund), og, i ~ 5% af tilfældene, har områder af osteoid (se fig. 1 i nedenstående Histopatologi af liposarkomer afsnit) eller bruskagtig materiale . Tumorerne viser også store variationer i deres celleindhold. For eksempel har op til 10% af DDL-tumorer områder med ALT/WDL-histopatologi, og sjældne tilfælde af DDL har områder, der indeholder meningotel-lignende hvirvler af flade celler.
Genetik
De neoplastiske celler i både DDL og ALT/WDL bærer lignende små supernumerære markørkromosomer (sSMC'er) og/eller gigantiske markørkromosomer, der indeholder ekstra dele af kromosom 12's arm på bånd 13 til 15. Dette kromosomale område omfatter to gener forbundet med tumorudvikling , MDM2- og CDK4 -generne. Tilstedeværelsen af ekstra kopier af disse to gener og/eller deres overproducerede proteinprodukter er en yderst følsom og specifik indikator for, at en lipomatøs tumor er en ALT/WDL eller DDL frem for en anden type lipomatøs tumor. Overekspression af MDM2- og CDK -generne og/eller andet genetisk materiale i sSMC'erne eller gigantiske markørkromosomer mistænkes for at fremme udvikling og/eller progression af DDL samt ALT/WDL -tumorer. Andre gener i sMMC og gigantisk markørkromosom, der også er overudtrykt i ALT/WDL og DDL neoplastiske celler, inkluderer HMGA2 , CPM , YEATS4 og DDIT3 . Sammenlignet med ALT/WDL neoplastiske celler, dog udtrykker DDL neoplastiske celler: 1) højere niveauer af generne i de to unormale kromosomer; dette kan bidrage til udviklingen af ALT/WDL til DDL; og 2) højere niveauer af genprodukter på den lange arm af kromosom 1 ved bånd 32, den lange arm af kromosom 6 ved bånd 33, og i ~ 25% af tilfældene, den korte arm af kromosom 1 ved bånd 32.2, som indeholder JUN -gen (dette gen er overudtrykt i DDL, men ikke ALT/WDL). Da JUN- genets produkt, c-jun , hæmmer celledød og fremmer cellespredning, kan dets overproduktion bidrage til progression af ALT/WDL til DDL og/eller malignitet af DDL-neoplastiske celler. Genekspressionsprofilering (dvs. måling af ekspressionen af produkterne af tusinder af gener fra celler, væv eller tumorer) har afsløret, at adipocyt -celledifferentiering og metaboliske veje i ALAT / WDL er opreguleres mens celleproliferation og DNA-beskadigelse respons veje opreguleres i DDL.
Diagnose
Den histopatologiske af DDL er ofte utilstrækkelig klar til at stille en fast diagnose. Imidlertid understøttes diagnosen DDL hos personer: hvis tumorer indeholder ALT/WDL blandet med DDL histologiske komponenter; med historier om at have en tidligere ALT/WDL; eller som præsenterer en retroperitoneal liposarcoma (DDL udgør ~ 57% af alle retroperitoneale liposarcomer). DDL -tumorer forekommer kun sjældent (<1% af tilfældene) som overfladiske hudtumorer; er næsten 5 gange mindre tilbøjelige end ALT/WDL til at forekomme i øjenhulen; og er ekstremt sjældne hos børn. Påvisning af tumorcelle -MDM2 -amplifikation er den diagnostiske guldstandard for at skelne WDL fra lipomer, dysplastiske lipomer, atypiske spindelcellesarkomer, pleomorfe lipomer og ensomme fibrøse tumorer. Alternativt understøtter detektion i tumorcellerne af et overudtrykt CDK4- gen eller tilstedeværelsen af enten de specifikke ALT/WDL-associerede sSMC'er eller kæmpe markørkromosom stærkt diagnosen DDL eller ALT/WDL. Den kliniske præsentation, histopatologi og genforskelle (f.eks. Tumorcelleoverekspression af cJUN -genet favoriserer stærkt diagnosen DDL frem for ATL/WDL) mellem de to sidstnævnte liposarkomformer hjælper normalt med at skelne mellem dem.
Behandling og prognose
Komplet kirurgisk resektion er normalt den anbefalede førstelinjebehandling for lokaliserede DDL-tumorer. Nye undersøgelser tyder imidlertid på, at patienter med DDL-tumorer, der er begrænset til en ekstremitet eller bagagerummet og har en forudsagt 10-årig tumorrelateret samlet overlevelse på 51% eller mindre, har forbedret resultater, når kemoterapi (f.eks. Doxorubicin plus ifosfamid ) tilføjes til deres kirurgiske behandlinger. For disse lokaliserede former for DDL kan periooperativ strålebehandling efter National Comprehensive Cancer Network -retningslinjer også overvejes.
Retroperitoneal DDL er den mest almindelige, kirurgisk uoverskuelige og alvorlige form for DDL: den har en tilbagefaldsprocent på 66% og en femårig samlet overlevelsesrate på 54%. Den primære behandlingsmulighed for retroperitoneal DDL er kirurgisk resektion. Et klinisk fase III -forsøg fandt ringe forskel i resultaterne af strålebehandling efterfulgt af kirurgisk resektion sammenlignet med kirurgisk resektion alene ved behandling af retroperitoneal DDL. I andre fase III kliniske forsøg blev DDL-patienter med utilgængelige retroperitoneale og/eller metastatiske tumorer behandlet med kemoterapi i frontlinjen, der sammenlignede doxorubicin med doxorubicin plus ifosfamid eller doxorubicin med gemcitabin plus docetaxel . Andre undersøgelser har ligeledes undersøgt værdien af forskellige kemoterapiregimer. Disse undersøgelser fandt ofte ringe forskel i de samlede overlevelsestider i deres sammenligninger, men viste nogle forbedringer i progressionsfri overlevelse og andre kliniske parametre. Baseret på disse undersøgelser er en anbefalet førstelinjebehandling for retroperitoneal og andre kirurgisk utilgængelige eller metastatiske DDL-tumorer behandling med et antracyklinbaseret kemoterapiregime eller, i tumorresistente eller tilbagefaldende tilfælde, eribulin kemoterapi. En gennemgang foretaget i 2020 rapporterede median overlevelsestider for lav histopatologisk grad og høj histopatologisk klasse DDL til henholdsvis 113 måneder og 48 måneder. Yderligere undersøgelser er nødvendige for at fremlægge bevis for effektiviteten af strålebehandling, kemoterapi og nye terapier i alle varianter af DDL.
Nye terapier
Flere nye terapiregimer for DDL og de mere aggressive eller på anden måde problematiske tilfælde af ALT/WDL er i øjeblikket under kliniske forsøg . Et klinisk fase II -studie, der undersøger abemaciclib, er i gang hos patienter med forbehandlet eller ubehandlet DDL. Foreløbig analyse viste, at denne hæmmer af CDK4- og CDK6- genernes produkt Serin/threonin-specifikke proteinkinaseenzymer producerede en forlænget median progressionsfri overlevelsestid på 30,4 uger. Et fase III multicenter, randomiseret, dobbeltblindet, placebokontrolleret klinisk studie af abemaciclib er i sin aktive fase og vil snart (som anført i juli 2021) begynde at rekruttere 108 personer med avanceret, tilbagevendende og/eller metastatisk DDL. Undersøgelsen er sponsoreret af Sarcoma Alliance for Research gennem samarbejde i samarbejde med Eli Lilly og Company . Ribociclib , også en CDK4- og CDK6 -geninhibitor , i kombination med en mTOR -hæmmer , er everolimus i et fase II klinisk forsøg med personer med avanceret DDL eller leiomyosarcoma . En fase III registreringsundersøgelse (dvs. en stor bekræftende undersøgelse beregnet til at etablere en acceptabel fordel/sikkerhedsprofil for at få lovgivningsmæssig godkendelse for en præcist defineret indikation) evaluerer sikkerheden og effekten af milademetan sammenlignet med trabectedin hos patienter med ikke -resekterbare (dvs. resektion anses for at forårsage uacceptabel morbiditet eller dødelighed) eller metastatisk DDL, der er udviklet på 1 eller flere tidligere systemiske behandlinger, herunder mindst 1 antracyklinbaseret terapi. Sponsoren, Rain Therapeutics Inc, rekrutterer i øjeblikket 160 personer til forsøget. Et andet fase III -klinisk forsøg undersøger MDM2 -hæmmer milademetan versus trabectedin , en blokering af den onkogene transkriptionsfaktor FUS -CHOP , ved MDM2 -overudtrykkende ALT/WDL og DDL. Milademetan har vist håndterbar toksicitet og en vis aktivitet, der resulterer i stabil sygdom og/eller et par delvise reaktioner i DDL.
Myxoid liposarkom
Præsentation
Myxoid liposarcoma (MLS), som omfatter en type liposarcom betegnet runde celle liposarcoma, repræsenterer ~ 30% af alle liposarcomer. Det har en højeste forekomst i individers fjerde og femte årti med en mandlig overvægt i de fleste undersøgelser. Selvom det er usædvanligt hos børn og unge, er MLS den mest almindelige liposarkomform, der diagnosticeres i disse aldersgrupper. MLS præsenterer typisk som en stor (1 til 39 cm; gennemsnit 12 cm), mobil, godt omskrevet, smertefri masse, der udviklede sig fra 1 uge til 15 år før diagnosen. MLS-tumorer er placeret i dybtliggende bløde væv i lårene (65-80% af tilfældene), underbenene (10-15% af tilfældene), retroperitoneum (8% af tilfældene) og arme (5% af tilfældene). I cirka en tredjedel af tilfældene metastaserer disse tumorer til andre steder af blødt væv (f.eks. Retroperitoneum, thorax eller anden ekstremitet), skeletben og/eller lunge. Enkeltpersoner kan opleve disse metastaser, især dem i knogler; det er blevet anbefalet, at patienterne testes ved præsentation for knoglemetastase ved medicinsk billeddannelse , herunder røntgenstråler , CT-scanninger og/eller magnetisk resonansbilleddannelse .
Patologi
Histopatologiske analyser af MLS (se figur 3 og 4 i nedenstående histopatologi for liposarkomer) afslører celler spredt gennem en myxoid matrix (dvs. en bindevævsbaggrund, der fremstår mere blå eller lilla end den normale farve af normalt bindevæv, når disse væv er korrekt forberedt, H&E farvet og set mikroskopisk). Disse celler er lipoblaster , hvoraf nogle er signetringformede (en form, der tyder på, at cellen kan være neoplastisk), ovalformet eller rundformet. MLS-tumorer kan være hypercellulære og indeholde solide plader af runde celler, der omfatter mindst 5% af alle celler eller lav cellularitet befolket med celler, der har intetsigende kerner og <5% runde celler i en baggrund af buede kapillærer, der ligner et kyllingetrådsmønster. Tumorer, der indeholder mindst 5% runde celler, klassificeres som højkvalitets, mens dem med <5% runde celler klassificeres som lavkvalitetsceller. Højkvalitets MLS-tumorer tager typisk et mere aggressivt klinisk forløb end lavgradige MLS-tumorer.
Genetik
MLS-tumorceller er praktisk talt defineret ved deres ekspression af et FUS-DDIT3- fusionsgen (også kaldet et kimært gen ), som forekommer i> 95% af tilfældene eller et EWSR1-DDIT3- fusionsgen, der forekommer i de resterende <5% af tilfældene. De FUS-DDIT3 fusionsgen- former som følge af en translokation (betegnet t (00:16) (q13: p11)) mellem det sted, hvor DDIT3 genet ved bånd 12 af kromosom 12 q arm og stedet for den FUS genet ved bånd 11 på kromosom 16's korte arm (også betegnet p -armen ). Den Fusionsproteinet (også betegnet kimært protein) produkt af denne kimære onkogen gen, FUS-DDIT3, er kendt for at standse fedt cellemodning og fremme neoplasi. Den EWSR1-DDIT3 fusionsgenet (betegnet t (12; 22) (q13; q12)) resultater fra en translokation af EWSR1 genet lokaliseret på bånd 12.2 på kromosom 22 q arm med DDIT2 genet. Fusionsproteinproduktet af EWSR1-DDIT3- genet fremmer , ligesom FUS-DDIT3-fusionsproteinet, neoplasi. På trods af disse fusionsgenrelationer kræves der yderligere undersøgelser for at definere deres bidrag til udvikling og/eller vedligeholdelse af MLS -tumorer.
Diagnose
Lavkvalitets- og mellemklasse MLS-tumorer kan identificeres histologisk ved deres klassiske morfologi med karakteristisk kyllingetrådsvaskulatur spredt gennem et myxoid stroma. Imidlertid kan MLS-tumorer af høj kvalitet være vanskelige at skelne fra andre runde celle-neoplasmer, især MLS-tumorer af høj kvalitet, der består af diffuse celle- og/eller ren runde cellemorfologi i en sådan grad, at dette klassiske kar-myxoid-mønster tilsløres. Påvisning af en DDIT3 gen omlejringer med FUS eller EWSR1 gen ved in situ hybridisering eller immunhistokemi eller RNA fusionstranskripter af disse gener ved realtid polymerasekædereaktioner bekræfter diagnosen af høj kvalitet samt tvetydige tilfælde af lav kvalitet eller mellemklasse MLS-tumorer.
Behandling og prognose
MLS er typisk blevet behandlet ved kirurgisk resektion, men kan kræve mere radikale indgreb, f.eks. Kan amputation af lemmer være nødvendig, når et lemmes neurovaskulære bundt er kompromitteret. Den postoperative risiko for tilbagefald inden for 3 år efter operationen er rapporteret til at være ~ 15%, når ikke alle tumorer fjernes og ~ 10%, når tumorfjernelsen er fuldført. Tilføjelsen af strålebehandling til kirurgisk resektion har forbedret den lokale kontrol af MLS -tumorer og er blevet anbefalet til behandling af ikke -resekterbar og tilbagevendende MLS. Imidlertid er der behov for yderligere undersøgelser for at bestemme værdien af strålebehandling ved behandling af de forskellige sorter af MLS. Kemoterapibehandlinger, der anvender ifosfamid , en anthracyclin, såsom daunorubicin , dacarbazin og/eller trabectedin, har vist sig at være nyttige: Et klinisk fase III-forsøg viste progressionsfrie overlevelsestider hos MLS-patienter behandlet med trabectedin eller dacarbazin til henholdsvis 5,6 og 1,5 måneder. I 2015 godkendte Food and Drug Administration trabectedin til brug i ikke -resekterbare og metastatiske liposarkomer.
Samlet set har den 10-årige overlevelsesrate for MLS-individer været 77%, en overlevelsesrate, der er betydeligt længere end andre liposarkomformer. Sammenlignet med lavrisiko MLS er højrisiko MLS (risiko defineret ved tumorrunde celleindhold og/eller andre ugunstige prognostiske indikatorer) forbundet med øgede metastaser og derfor en kortere overlevelsestid. Øget tumorstørrelse (≥ 10 cm) er stærkt forbundet med MLS af højere kvalitet og derfor en kortere overlevelsestid. Andre faktorer, der har været forbundet med ugunstige resultater i MLS, omfatter tilstedeværelse af tumornekrose, alder> 45 år, P53 -genoverekspression og mandligt køn. Den runde celleform af myxoide liposarcomer ser også ud til at have en relativt dårlig prognose: i forskellige retrospektive anmeldelser blev myxoid liposarcom normalt fundet at være lavgradig og derfor relativt lydhør over for kemoterapi, hvorimod myxoid liposarkom af høj kvalitet (dvs. myxoid liposarkom) havde højere satser af metastase, opførte sig mere aggressivt og reagerede ikke godt på kemoterapi.Det er dog vigtigt at bemærke, at næsten alle tilfælde af myxoide liposarkomer hos pædiatriske patienter har haft fremragende prognoser.
Nye terapier
En PPAR-y- agonist (dvs. aktivator), efatutazon, blev undersøgt i et lille fase I-forsøg på personer med forskellige maligniteter i avanceret stadium. Lægemidlet producerede en markant holdbar reaktion hos en person med MLS, hvilket tyder på, at PPAR-y-agonister ville være nyttige til behandling af denne sygdom. Et klinisk fase II-forsøg udført i Italien undersøger virkningerne af et trabectedin plus pioglitazon (en anden PPAR-γ-agonist) hos personer med stabile MLS-tumorer. Undersøgelsen involverer to sekventielle trin. Det første trin undersøger responsen fra patienter behandlet i mindst 4 cyklusser med trabectedin alene. Hvis der opnås stabil sygdom, vil det andet trin undersøge virkningerne af yderligere behandling af initialt reagerende patienter med en kombination af trabectedin og pioglitazon. Et klinisk fase II -forsøg er ved at være afsluttet for at evaluere effekten af sirolimus (en hæmmer af MTOR ; sirolimus er også kendt som rapamycin) plus cyclophosphamid (et kemoterapimedicin) i metastatisk eller ikke -resekterbart MLS. Et klinisk fase II-forsøg rekrutterer patienter til at evaluere sintilimab (et humant monoklonalt IgG4 -antistof rettet mod det programmerede celledødsprotein 1 placeret på overfladen af celler) i kombination med to kemoterapimediciner, doxorubicin og ifosfamid , som førstelinjebehandling af blød vævsarkomer inklusive MLS.
T-celler er blevet genetisk manipuleret til at målrette mod MAGE-A4- antigenet udtrykt på et HLA-A*02 MAGE-A4-holdigt peptid placeret på overfladen af de neoplastiske celler i visse typer tumorer. Disse manipulerede celler (betegnet ADP-A2M4-T-celler) angreb og dræbte forskellige dyrkede humane kræftceller, der bærer dette antigen, og i en klinisk fase 1-undersøgelse krympet forskellige solide tumortyper hos patienter, hvis tumorer indeholdt neoplastiske celler, der udtrykker dette antigen. Et klinisk fase II-studie rekrutterer nu individer til at undersøge effektiviteten og sikkerheden af ADP-A2M4 T-celler (konstrueret fra modtagerens egne T-celler) hos HLA-A*02-postitive patienter med metastatisk eller uoperabel, avanceret fase MSGE-4 -positive MLS -tumorer.
Pleomorf liposarkom
Præsentation
Pleomorfe liposarkomer (PLS), der tegner sig for 5% til 10% af alle liposarkom-tilfælde, er hurtigt voksende, normalt store (> 5 cm) og smertefrie, men stærkt maligne adipocyttumorer. De forekommer primært hos personer> 50 år med en overvægt hos kvinder. PLS -tumorer findes sjældent hos børn. PLS -tumorer findes i et ben eller en arm (65% af tilfældene), retroperitoneum eller underlivet (15% af tilfældene), eller i sjældne tilfælde stammen, sædstrengen , hoved- og nakkeområder, brystvæg, bækkenhulrum , lungebetændelse , perikard og rygsøjle. Disse tumorer er normalt lokaliseret i dybe bløde væv med kun 25% af tilfældene i subkutant væv. Sjældne tilfælde af PLS har vist sig hos personer med Li-Fraumeni eller Muir-Torre syndromer , to arvelige genetiske lidelser, der prædisponerer ramte for at udvikle forskellige former for kræft.
Patologi
Histopatologien for PLS -tumorer består ofte af områder, der ligner myxoid liposarcom blandet med områder, der indeholder udifferentierede celler. Disse tumorer er markeret hypercellulære og indeholder mindst nogle variabelt formede lipoblaster, der har pleomorfe kerner. Nekroseområder er almindelige, gigantiske celler, hvoraf nogle er flerkerne og/eller indeholder opslugte neutrofiler , er lejlighedsvis til stede, og hyalindråber kan ses i nogle celler såvel som spredt ekstracellulært i hele tumoren. Den udifferentierede komponent i disse tumorer består oftest af spindelformede celler, hvor 25% af tilfældene viser celler med en epitelioide cellemorfologi. Disse tumorer har i det mindste nogle foci med en histopatologi svarende til histiocytomer af myxofibrosarcoma af høj kvalitet , en tumor, der tidligere blev betegnet malign myxoid fibrous histiocytoma.
Genetik
PLS -neoplastiske celler indeholder forskellige gen- og kromosomafvigelser: TP53 -genet slettes eller muteres i 17–60% af tilfældene; den RB1 genet er deleteret i 60% af tilfældene; og Neurofibromin 1 -genet går tabt ved inaktivering af mutationer i 8% af tilfældene eller i sjældnere tilfælde ved en deletion omkring dets placering i bånd 11.2 på den lange arm af kromosom 12. Disse celler kan også vise gevinster i det genetiske materiale omkring: bånd 12 –15 på den korte arm af kromosom 5; bånd 21 på den korte arm af kromosom 1; og bånd 22 på kromosomets lange arm 7. Ændringerne i antallet af genkopier induceret af disse abnormiteter ligner dem, der ses i myxofibrosarcoma -typen af histiocytomerne . Rollen (erne) for disse ændringer i genkopienumre ved fremme af PLS er ikke blevet defineret. PLS er således ulig andre liposarkomer, idet dets neoplastiske celler har et komplekst genom uden karakteristiske genomiske ændringer eller identificerbare gener, der driver sygdommen. Påvisning af ændringer i ekspressionen af TP53-, RB1- og neurofibromin 1- generne samt andre, mindre almindeligt ændrede gener i PLS (f.eks. PIK3CA , tyrosin-proteinkinase SYK , PTK2B , EPHA5 og ERBB4 ) kan hjælpe med at understøtte definerer ikke klart en tumor som værende PLS. Udvidelse af kromosom telomere ender ved patologiske mekanismer betegnes alternativ forlængelse af telomerer forekommer i de neoplastiske celler af ~ 80% af PLS tilfælde, men er langt mindre udbredt eller ikke set i de andre fire former for liposarcom.
Diagnose
Diagnosen PLS afhænger af dens præsentation, histopatologi og genetik. Histopatologien for PLS ligner ofte myxofibrosarcoma, men adskiller sig fra denne tumor ved dets indhold af pleomorfe lipoblaster.
Behandling og prognose
Radikal kirurgisk resektion er den vigtigste behandling for PLS; det er også en vigtig palliativ intervention for at lindre symptomer på grund af komprimering af organer og væv. Kirurgi kan kræve fjernelse af et helt komprimeret organ, såsom nyren eller tyktarmen. Uanset denne operation er lokale tilbagefaldshastigheder imidlertid meget høje. Anvendelser af kemoterapi og/eller strålebehandling i forbindelse med radikal kirurgi har ikke vist sig at forlænge overlevelsen og betragtes som kontroversielle indgreb. Den Nationale Comprehensive Cancer Network anbefaler behandling for personer med høj risiko lokaliserede PLS ved komplet kirurgisk resektion, når det er muligt, kombineret med strålebehandling. Personer med metastatisk sygdom er blevet behandlet med kemoterapi (f.eks. Doxorubicin plus ifosfamid eller eribulin ) svarende til de regimer, der anvendes til dedifferentieret liposarkom (se ovenstående afsnit om behandling af denne liposarkom) Omkring 20% af PLS -tumorer metastaserer til fjerne steder, mest fælles heraf er lunge (82% af metastaser), lever (18% af metastaser) og knogler eller bugspytkirtel (18% af metastaser). PLS overlevelsesrater efter 1, 3 og 5 år rapporteres at være henholdsvis 93%, 75%og 29%. Tumorer placeret i bagagerumets midterposition, større end 10 cm i størrelse, dybt siddende eller indeholdende områder med nekrose har dårligere prognoser.
Myxoid pleomorf liposarkom
Myxoid pleomorf liposarcoma (oprindeligt betegnet pleomorft myxoid liposarcoma) blev først beskrevet i en stor undersøgelse af liposarkomerne i 2009. Selvom det oprindeligt blev betragtet som en mulig variant af de myxoide liposarkomer med pleomorfe træk, klassificerede Verdenssundhedsorganisationen (2020) det som en ny og tydelig form for liposarkomerne. Denne klassifikation var baseret på fund om, at de myxoide pleomorfe liposarkomer, selvom de havde histopatologiske træk, der lignede myxoide liposarkomer, havde kliniske og vigtigst af alt kritiske genetiske og molekylære egenskaber, der adskilte sig fra myxoide såvel som de andre tre liposarkomformer.
Præsentation
Myxoid pleomorf liposarcoma (MPL) er en usædvanligt sjælden og meget aggressiv form for liposarkomerne, der udvikler sig hos børn, unge, unge voksne og i en nyere undersøgelse individer> 50 år. MPL-tumorer findes som dybe bløde vævsmasser, der ofte er placeret i mediastinum og, sjældnere, ekstremiteterne, hovedet og nakken, bughulen eller bagagerummet. Mindst to tilfælde af MPL har vist sig individuelt med Li - Fraumeni syndrom , en arvelig genetisk lidelse, der disponerer individer for at udvikle forskellige kræftformer.
Patologi
Ved histopatologiske analyser består MPL -tumorer af områder, der ligner konventionel myxoid liposarkom; disse områder, der repræsenterer 30-50% af de samlede tumorområder, har en rigelig myxoid matrix, en veludviklet kapillær vaskulatur, intetsigende celler, der er runde og/eller let spindelformede, vakuolerede lipoblaster og flerkernede celler formet som små blomster. Disse områder indeholder imidlertid også en spredning af stærkt pleomorfe celler, der viser større grader af atomforstørrelse og uregelmæssighed end de celler, der har set myxoide liposarkomtumorer. Andre områder af MPL -tumorer er mere cellulære og består af hurtigt voksende og stærkt pleomorfe lipoblaster .
Genetik
De neoplastiske celler i MPL udtrykker ikke FUS-DDIT3- eller EWSR1-DDIT3- fusionsgenerne, der udtrykkes af de neoplastiske celler i henholdsvis> 95% eller <5% af myxoid fibrosarcoma-tilfælde. Inaktivering af RB1 tumorsuppressorgen grund af sin deletion eller patologisk undertrykkelse findes i alle tilfælde MPL. MPL -neoplastiske celler har også almindeligvis andre ændringer i deres kromosomer. De kan vise unormale gevinster i nogle af det genetiske materiale, der normalt findes på kromosomer 1, 6, 7, 8, 19, 21 og/eller X og tab i det genetiske materiale, der normalt findes på kromosomer 2, 3, 4, 5, 10 , 11, 12, 13, 14, 15, 16, 17 og / eller 22. det genetiske materiale tabt i bånd 14 på den lange arm af kromosom 13 omfatter ikke blot RP1 gen, men også RCBTB2 , DLEU1 , og ITM2B gener. På grund af dens sjældenhed og nyere definition mangler de molekylære egenskaber og betydningen af disse genetiske abnormiteter endnu ikke at være fuldstændigt defineret. Ikke desto mindre har undersøgelser antydet, at tab i et eller flere af RB1-, RCBTB2-, DLEU1- og ITM2B -generne , men især RP1 -genet, kan være involveret i at bidrage til udviklingen og/eller progressionen af MPL.
Diagnose
Diagnosen MPL afhænger af dens tumors kliniske præsentation, histopatologiske lighed med myxoid liposarkom og mest kritisk fravær af FUS-DDIT3 sn EWSR1-DDIT3 fusionsgener i dets neoplastiske celler.
Behandling og prognose
Mens personer med MPL er blevet behandlet med kirurgisk resektion for at fjerne deres tumorer, fandt en gennemgang i 2021, at der ikke var konsensusanbefalinger for standarden for pleje af MPL med hensyn til stråle- og kemoterapiregimer (når de bruges enten alene eller kombineret med kirurgi) for behandling af disse tumorer.
Histopatologi af liposarkomer
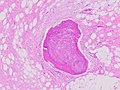
Fig. 1 Mikrograf af knogledannelse i en liposarcoma -tumor

Fig. 2 Mikrograf af en dedifferentieret liposarkom -tumor

Fig. 3 Mikrograf med lavere effekt af myxoid liposarkomtumor

Fig. 4 Mikrograf med større effekt af myxoid liposarkomtumor


.jpg)
.JPG)
Medicinsk billeddannelse
Medicinsk ultralyd og magnetisk resonansbilleddannelse (MRI) af liposarkomer er nyttige og ofte afgørende for at bestemme deres omfang, kirurgisk tilgængelighed og forhold til eventuelle observerede organdysfunktioner. Da ultralydsundersøgelse sædvanligvis ikke er i stand til at skelne et liposarkom fra et godartet lipom, er MR den indledende billeddannelse, der er valgt som bevis for bevis for, at denne adskiller sig.

Fig. 5 Ultrasonografi af et liposarkom med områder med højt ekko reflekteret fra dets lipomatøse matrix og områder med lavt ekko reflekteret fra dets ikke-lipomatiske områder.
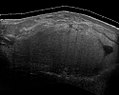
Fig. 6 Ultrasonografi af et liposarkom, der efterligner et lipom. Denne homogene høj-ekkoiske masse har samme udseende som et lipoma.

Fig. 7 MR af myxoid liposarkom af høj kvalitet, i den venstre aksillære region af en 40 -årig mand, fremhævet af dens hvide farve, i dette vandrette snit af tumoren.



Samfund og kultur
Bemærkelsesværdige sager
- Chad Brown (1961–2014), en pokerspiller, døde af liposarkom
- Richard Feynman (1918–1988), en teoretisk fysiker, døde efter en operation for at behandle sygdommen.
- Rob Ford (1969–2016), tidligere borgmester i Toronto og byrådsmedlem i Toronto, døde af pleomorf liposarkom.
- Hokie Gajan (1959–2016), tidligere løbende tilbage for New Orleans Saints og radiofarvekommentator for holdet, døde af liposarkom.
- Charlie Davies (1986-), tidligere fodboldspiller for Philadelphia Union of Major League Soccer, fik diagnosen liposarkom i 2016.
- Mark Strand (1934-2014), tidligere amerikansk digter og Pulitzer-prisvinder, døde af liposarkom.
Se også
- Lipoma
- Wendy Walk , non-profit organisation, hvis mission er at skaffe midler og bevidsthed om sarkomer, herunder liposarkom
Referencer
eksterne links
| Klassifikation | |
|---|---|
| Eksterne ressourcer |